2 Prepare data
In this chapter, we describe how to prepare two different input datasets, where the annotation part of VCFshiny can only input unannotated VCF files, and the analysis part of VCFshiny can read annotated VCF and TXT files.
2.1 Source of VCF input data
The Variant Call Format (VCF) is used to record gene sequence variations. It is also the first file format to be understood for genome population correlation analysis. The file is divided into two main parts: the Header comment section, which begins with #, and the body section.
2.2 Source of TXT input data
TXT file is one of the standard storage formats after mutation data annotation. Because some users cannot annotate the steps, We provide Annovar (Wang K, Li M, Hakonarson H. 2010) and VariantAnnotation (Obenchain, V., et al. 2014) two methods of annotation in the VCFshiny, Users can choose to annotate themselves or use the tool.
2.3 Input Data name requirements
2.3.1 Input file name requirements

Figure 2.1: File-Format
- The first box represents the sample name, which can be the group of experiments and the number of repetitions, connected by the character
"-"or"_". - The second box represents the data type, which can be snp or indel data. When snp and indel are not classified in the data, this box is not necessary.
- The third box represents the data format, which can be vcf files, vcf. gz compressed files, and Annovar annotated TXT files.
- The contents of the three boxes are connected by
".".
2.3.2 Input compress files requirements
Before uploading the data to VCFshiny, that needs to be compressed. The following are the naming requirements for the compressed folder.
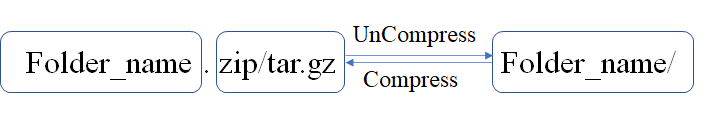
Figure 2.2: Fold_Format
- The compressed file name must be the same as the name of the compressed folder.
- The compressed file can be in
*.tar.gzor*.zipformat.
2.4 Example data set in VCFshiny software
We provide a built-in dataset that can be used to explore VCFshiny. The data set can be directly loaded into the APP by clicking the button “Use example data?” of the data input module (Figure S4-II). The built-in dataset was derived from the sequencing data of published articles, including a control group and three experimental groups, with three replicates in each group (Liang Y, Xie J, Zhang Q, et al. 2022). The sequencing data was first mapping to the reference, followed by gatk Variants calling and Annovar annotations.